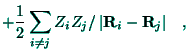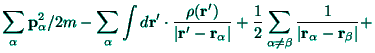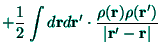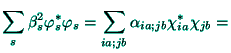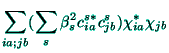Beside the atomic-like orbitals (56) the molecular-like
orbitals[11] ![]() include extended bond-like orbitals
include extended bond-like orbitals ![]() , such that
, such that
The great disparity between the scale-lengths of the localized
atomic-like orbitals ![]() and the extended bond-like orbitals
and the extended bond-like orbitals ![]() provides a certain decoupling of the atomic degrees of freedom
from the chemical bond degrees of freedom, up to a density-density
interaction originating in the direct Coulomb repulsion; the minimization of
the Hartree-Foch energy functional for the molecular-like orbitals
provides a certain decoupling of the atomic degrees of freedom
from the chemical bond degrees of freedom, up to a density-density
interaction originating in the direct Coulomb repulsion; the minimization of
the Hartree-Foch energy functional for the molecular-like orbitals ![]() with respect to the
with respect to the ![]() -parameters leads to the linear system of
equations[14]
-parameters leads to the linear system of
equations[14]
Beside the atomic-like energy functional ![]() given by (57) one obtains now an additional bond-like energy functional
given by (57) one obtains now an additional bond-like energy functional
The atomic-like problem as formulated by the energy functional ![]() given by (57) can be solved, in principle, according to the
usual practice of the wavefunctions methods;[10] the bond-like
hamiltonian
given by (57) can be solved, in principle, according to the
usual practice of the wavefunctions methods;[10] the bond-like
hamiltonian ![]() given by (69) can be treated by the
quasi-classical theory of the slightly inhomogeneous electron liquid; the
self-consistent solution to equations (63) is then obtained for the
parameters
given by (69) can be treated by the
quasi-classical theory of the slightly inhomogeneous electron liquid; the
self-consistent solution to equations (63) is then obtained for the
parameters
![]() and, implicitly, for the density
and, implicitly, for the density ![]() of the
effective charge of the ionic cores; the actual self-consistent solution
must also ensure the minimum value of the total energy functional
of the
effective charge of the ionic cores; the actual self-consistent solution
must also ensure the minimum value of the total energy functional
![]() with respect to the positions of the atomic nuclei.
The chemical bond consists therefore of extended bond-like electronic
orbitals of fractional occupancy
with respect to the positions of the atomic nuclei.
The chemical bond consists therefore of extended bond-like electronic
orbitals of fractional occupancy
![]() and of localized
atomic-like electronic orbitals of fractional occupancy
and of localized
atomic-like electronic orbitals of fractional occupancy
![]() (the atomic-like fractional occupancy may be made to appear explicitly in
the atomic-like energy functional
(the atomic-like fractional occupancy may be made to appear explicitly in
the atomic-like energy functional ![]() ); the total electronic energy
ensures the equilibrium of the atomic aggregate with respect to the Coulomb
repulsion between the atomic nuclei. The contribution of the atomic-like
part of the total energy to the binding energy is a quantal correction with
respect to the quasi-classical description, so that, to the first
approximation it may be neglected; the unrestricted minimization of the
bond-like energy functional requires then a unity occupancy
); the total electronic energy
ensures the equilibrium of the atomic aggregate with respect to the Coulomb
repulsion between the atomic nuclei. The contribution of the atomic-like
part of the total energy to the binding energy is a quantal correction with
respect to the quasi-classical description, so that, to the first
approximation it may be neglected; the unrestricted minimization of the
bond-like energy functional requires then a unity occupancy
![]() for the bond-like orbitals, the conservation of the total
charge being ensured by the Coulomb interactions in the hamiltonian
for the bond-like orbitals, the conservation of the total
charge being ensured by the Coulomb interactions in the hamiltonian ![]() given by (69); within this approximation, one may say, therefore, that
the binding energy and the cohesion of the atomic aggregates are given by
the theory of the slightly inhomogeneous electron liquid for the bond-like
hamiltonian
given by (69); within this approximation, one may say, therefore, that
the binding energy and the cohesion of the atomic aggregates are given by
the theory of the slightly inhomogeneous electron liquid for the bond-like
hamiltonian ![]() given by (69); however, while this procedure is
satisfactory for the binding energy and the cohesion, the fractional
occupancy must be included for the single-electron properties.
given by (69); however, while this procedure is
satisfactory for the binding energy and the cohesion, the fractional
occupancy must be included for the single-electron properties.